
fr.wedoany.com Rapport : Des chercheurs de l'Université de Tohoku (Tohoku University) ont découvert un nouveau principe de conception de catalyseurs, révélant que les catalyseurs à double atome (dual-atom catalysts, DACs) présentent un mode « double optimum de Sabatier » (dual-Sabatier optima) dans la réaction de réduction de l'oxygène, remettant en cause l'hypothèse du modèle volcanique unimodal utilisée depuis des décennies, et ouvrant potentiellement une nouvelle voie pour réduire le coût des piles à combustible à hydrogène.
Les piles à combustible sont considérées comme des dispositifs clés pour la construction d'une société à faible émission de carbone, car elles produisent de l'électricité à partir d'hydrogène avec un rejet propre. Cependant, de nombreuses piles à combustible dépendent encore de métaux précieux comme le platine pour piloter la réaction de réduction de l'oxygène (ORR), un processus qui affecte directement les performances et le coût. La théorie catalytique traditionnelle explique l'activité par le modèle « volcan unimodal », selon lequel le meilleur catalyseur se situe dans une plage étroite de propriétés chimiques. Pourtant, en analysant un vaste ensemble de données expérimentales issues de la plateforme de catalyse numérique (Digital Catalysis Platform, DigCat), l'équipe de recherche a constaté que les catalyseurs à double atome ne suivaient pas ce modèle attendu.
Les chercheurs ont utilisé des simulations théoriques avancées, la modélisation microcinétique (microkinetic modeling) et l'apprentissage automatique pour étudier plus de 200 catalyseurs à double atome. Les résultats montrent que les DACs sont principalement contrôlés par une voie réactionnelle appelée mécanisme dissociatif (dissociative mechanism), contrairement au mécanisme associatif (associative mechanism) courant dans les catalyseurs à atome unique. Ce changement a un impact majeur sur l'activité catalytique : les DACs ne présentent plus un seul pic de performance optimale, mais deux zones optimales distinctes, soit le « double optimum de Sabatier ». L'apparition de ces deux pics résulte du transfert de l'étape limitante au cours de la réaction, qui alterne entre la dissociation de l'oxygène, la protonation de l'oxygène et la protonation de l'hydroxyle.
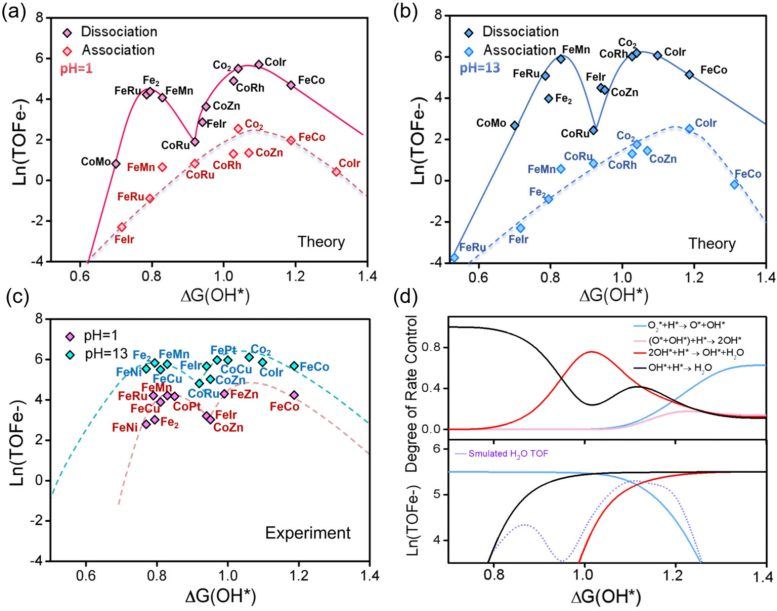
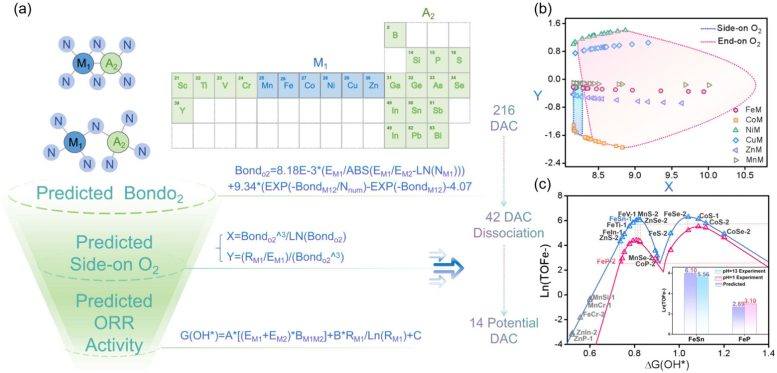
Les chercheurs ont découvert que ce principe s'applique à divers types de catalyseurs, incluant des systèmes composés de métaux de transition, d'éléments métalloïdes, voire d'atomes non métalliques. En combinant l'apprentissage automatique interprétable (interpretable machine learning) avec la modélisation théorique, l'équipe a construit un cadre prédictif capable d'identifier rapidement des structures catalytiques prometteuses. Hao Li, professeur émérite à l'Institut de recherche sur les matériaux avancés (WPI-AIMR) de l'Université de Tohoku, a déclaré que l'hypothèse longtemps admise selon laquelle les catalyseurs à double atome suivent les mêmes règles d'activité que les catalyseurs à atome unique est remise en cause : ces travaux montrent que lorsque deux atomes coopèrent, des mécanismes totalement différents peuvent émerger, ouvrant de nouvelles opportunités pour concevoir des matériaux efficaces destinés aux technologies d'énergie propre.
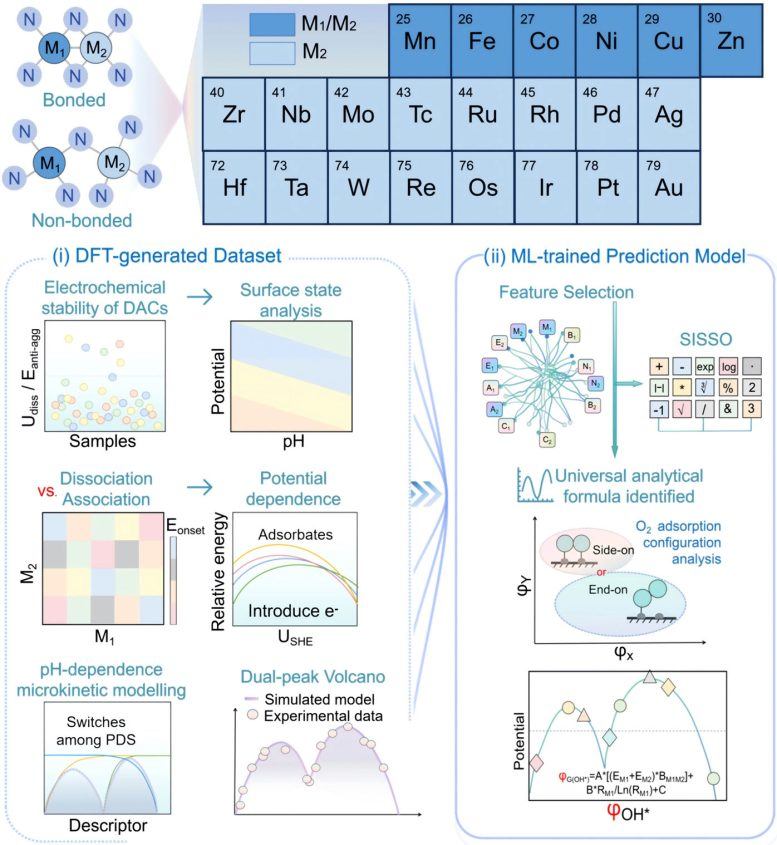
L'impact potentiel de cette découverte pourrait dépasser le cadre des piles à combustible et guider le développement de catalyseurs pour d'autres processus de conversion d'énergie et de production chimique. L'étude montre également comment l'intelligence artificielle peut extraire des lois scientifiques cachées à partir de données expérimentales existantes, accélérant ainsi le criblage de nouveaux matériaux. Prochainement, l'équipe prévoit d'appliquer cette méthode à des catalyseurs multimétalliques plus complexes, ainsi qu'à d'autres réactions liées à l'énergie en dehors de l'ORR, et de créer un système numérique entièrement autonome pour la conception rapide de catalyseurs de nouvelle génération destinés à l'énergie durable, en intégrant des agents d'IA, l'apprentissage automatique et la simulation électrochimique dans la plateforme DigCat.










